
电子与通信工程学院、人工智能学院教师孙畅以第一作者的身份在期刊Journal of Chemical Information and Modeling上发表论文“DCGCN: Dual-channel Graph Convolutional Network-based Drug-target Interactions Prediction Method with 3D Molecular Structure”。
探索药物-靶标相互作用(DTIs)对药物发现至关重要。目前大多数预测DTI的方法仅依赖于分子的线性结构,如SMILES或氨基酸序列。然而,这些线性特征未能充分反映分子的子结构或原子的相对位置。尽管出现了一些利用二维分子结构(如分子的键线式或原子图)的工作,但它们依然无法全面反映分子的化学结构。为此,本文提出了一种基于三维分子结构的DTI预测方法——DCGCN。DCGCN将分子的三维点云数据分解为三个组成部分:原子序列、原子连接性和距离图。这样的处理不仅有利于模型捕捉药物和靶标的分子组成,还有利于学习原子或氨基酸残基之间的连接性和距离。
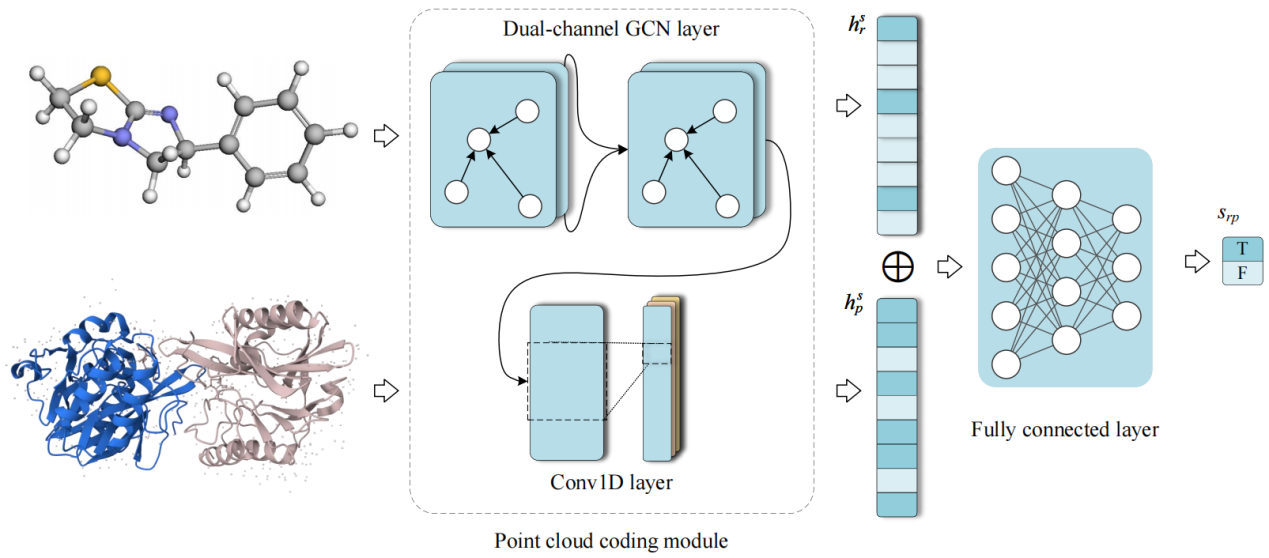
图1 DCGCN算法流程图
在两个公共数据集上的实验结果表明,DCGCN在整体性能上优于几种先进的DTI预测方法。这一优越性可归因于三维分子结构比线性结构更全面地反映和定义分子的特性。消融实验的结果表明,三维结构比线性结构提供了更有价值的见解。此外,DCGCN在冷启动设置下的优越性能进一步验证了其在识别新药物和靶标新用途方面的有效性。
表1 DCGCN算法性能对比

案例研究显示,DCGCN能够利用已知的DTI,通过将候选药物的化学结构和相互作用模式与已知配体进行比较,识别潜在的DTI。因此,DCGCN成为了一种实用工具,帮助生物学家筛选可靠的药物-靶标候选,提高生物识别实验的工作效率,加速药物开发。
天津师范大学电子与通信工程学院、人工智能学院教师孙畅为论文第一作者,天津师范大学为第一单位。该项研究由天津师范大学、天津市五中心医院的科研人员共同合作完成。该研究得到了国家自然科学基金青年科学基金项目(62401392)的支持。
文章地址:https://pubs.acs.org/doi/full/10.1021/acs.jcim.5c01012



